
November 17, 2018
I.
It is one of this country's great scientific achievements.
The first drug ever approved that can fix a faulty gene.
It's called Glybera, and it can treat a painful and potentially deadly genetic disorder with a single dose — a genuine made-in-Canada medical breakthrough.
But most Canadians have never heard of it.
A team of researchers at the University of British Columbia spent decades developing the treatment for people born with a genetic mutation that causes lipoprotein lipase defficiency (LPLD).
LPLD affects communities in the Saguenay region of northeastern Quebec at a higher rate than anywhere else in the world.
Montreal psychologist Cynthia Turcotte, 42, was born with LPLD, but wasn't diagnosed until it almost killed her when she was eight months old.
As a result of the gene mutation, her body is missing an essential protein that processes dietary fat. Her blood becomes thick and white with fat particles that can destroy her pancreas.
All her life, Turcotte has had to follow a strict diet. She can't eat cheese or chocolate or any food that contains fat. And she can't drink even a drop of alcohol.
All of that made it difficult to have a normal social life when she was younger.
But the worst part was learning that she could never have children. Women with the disease are warned to avoid pregnancy because there is a high risk of miscarriage.
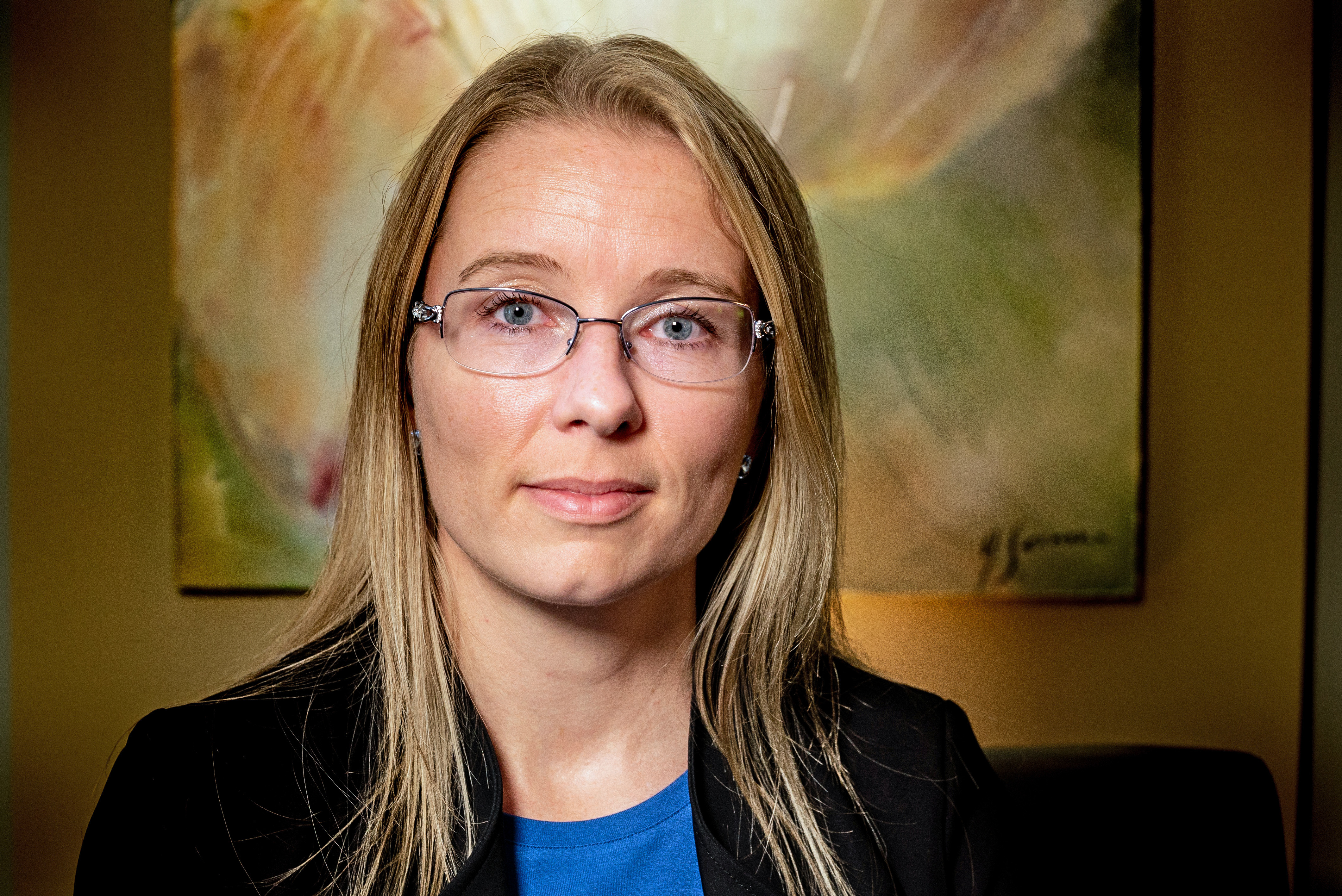
Despite accepting all of that and following a very strict diet, Turcotte experienced the most dangerous symptom of LPLD a decade ago — an agonizing pancreatitis attack.
"It's like digesting yourself," she said of the pain.
"Pancreatitis is like acid going through your belly and your abdomen. At that time, I was screaming. I was hospitalized for 10 days."
Frightened for her future, Turcotte learned about a clinical trial in Chicoutimi, Que., where doctors were testing an experimental treatment for LPLD. She immediately volunteered.
A series of injections in her leg muscles during a single visit changed Turcotte's life, ending the pancreatitis attacks and giving her the chance to start the family she thought she’d never have.
"It was a turning point in my life," said Turcotte, who is the proud mother of two young children — her "two miracles," she calls them.
But Turcotte would be one of only 31 people with LPLD to ever experience the benefits of Glybera.
The drug works. It is safe. But it's no longer available anywhere in the world.

Glybera was never sold in North America and was available in Europe for just two years, beginning in 2015. During that time, only one patient received the drug. Then it was abandoned by the company that held its European licensing rights.
The problem was the price.
The world's first gene therapy, a remarkable discovery by a dedicated team of scientists who came together in a Vancouver lab, had earned a second, more dubious distinction:
The world's most expensive drug.
II.
Dr. Michael Hayden has spent decades at UBC and the BC Children's Hospital exploring the mysteries of human genetics in search of cures for some of the most devastating inherited diseases.
If you can catch up with him on a break in his busy schedule and ask him about Glybera, his face lights up. Not many scientists can claim to have discovered how to fix a flaw in a human gene.
Hayden is also a medical doctor. One of his specialties is lipid disorders — disruptions in the body’s fat metabolism. So he's seen many patients suffering from LPLD.
It's a disease that affects about two or three people out of a million. But in the Saguenay region of Quebec, the rate can be up to 200 people out of a million. Geneticists call it "the founder effect," where a gene mutation from a single ancestor gets passed down through generations.
Doctors have tried every fat and cholesterol-lowering drug on the shelf to treat LPLD, but nothing works.

All their lives, patients must be vigilant in avoiding dietary fats — even a momentary splurge can have potentially deadly consequences. And no matter how strictly they control their fat intake, they can still suffer debilitating and potentially deadly bouts of pancreatitis.
For decades, no one knew for sure what was causing LPLD, although scientists suspected a genetic mutation. To prove it, they first needed to find the gene.
In 1986, Hayden assigned that challenge to a young doctor from Amsterdam named John Kastelein, who had joined his lab in Vancouver.
At the time, genetic research was in its infancy. It was decades before gene sequencing became routine.
Using the DNA of a 19-year-old man with LPLD, Kastelein and his colleagues searched for evidence a faulty gene was causing the patient's low levels of lipoprotein lipase.
The genetic research tools at the time were relatively primitive, so it was fortunate that the young man's mutation was unusually prominent.
"If it wasn't this bizarre hole in a gene we would not have found it at that time," Kastelein said. "This was a very big deal."

But finding the defective gene was just the first challenge. The next was to figure out a way to repair it.
Hayden's idea was simple and elegant, at least in theory: Just replace the defective gene with a new gene.
But how to deliver it to the right place in the body?
The team's answer was to use a harmless virus specially designed to carry a human gene. The virus would insert itself into the cell and make copies of the new gene.
The new genes would start producing the missing protein, which would then clear the fat from the blood and improve the health of the LPLD patient.
The solution was easy enough to imagine. But in order to demonstrate it could actually work, Hayden invited another young scientist to join his team.
It felt to me like we were headed off to climb Everest.
Dr. Colin Ross has been thinking about curing genetic diseases since he was a child.
That's when he first saw the effects of a genetic disease on a young classmate. The boy had inherited fragile X syndrome, which causes a range of physical and intellectual impairments, including attention disorders.
"He was a really great kid and they were trying to have him fit into the classroom. In the end, it didn’t work, but it taught me about this whole area I didn’t know about."
With the seed planted in elementary school, Ross pursued a career in genetics. By the time he finished his PhD at McMaster University in Hamilton in 2000, the human genome had been sequenced, and the possibility of repairing faulty genes seemed within reach.

He'd heard about the gene therapy work happening in Vancouver and wrote to Michael Hayden asking for a position in his lab.
Hayden agreed, and Ross moved to Vancouver and soon began testing the experimental gene therapy in a strain of mice specially engineered to carry the LPLD mutation.
"It felt to me like we were headed off to climb Everest," Ross said. "It seemed an impossible task, but I was optimistic that it could be done."
Animal research is an essential part of all drug development. The first animal used for a test is usually a mouse that has been genetically modified to exhibit the same genetic defects as the human disease.
The problem for Hayden's team was LPLD can kill a mouse within 24 hours of birth.
But if they could keep the mice alive, it would be a clear sign the treatment might ultimately work in humans.
Ross still remembers the pivotal experiment in Hayden's lab on Jan. 10, 2002.
"If it didn’t work this day, I didn't know what else we were going to do," he said. "It was a stressful time."
He didn't have to wait long for the result.
"As soon as I took a blood sample, you could see they didn't have this white blood anymore. It was nice, normal-looking blood. It was incredible. It was just beautiful."
The stunning results of the mouse experiments were featured on the cover of the journal Human Gene Therapy in September 2004. The cover image showed how, week by week, the mouse blood changed from milky white to a clear, transparent red, illustrating just how effectively the gene therapy was working.
"That was an amazing moment," Hayden said. "You knew you had it then."

But before they could move to human trials, Hayden’s team needed to test the virus in a larger mammal.
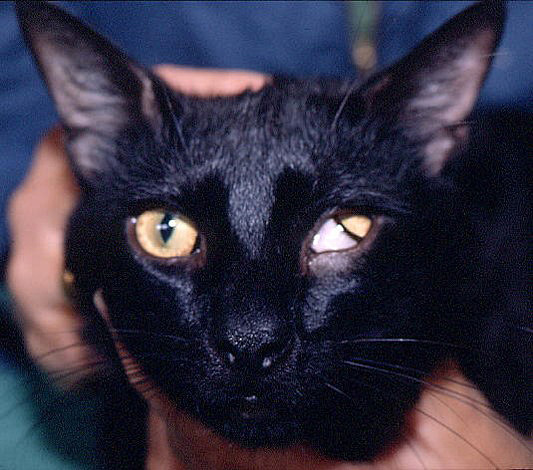
Serendipity struck again, this time at a lipid conference in Florence, Italy, where Hayden met Boyce Jones, a veterinarian from New Zealand who had discovered a curious family of cats with white blood.
Jones agreed to help Hayden, first by shipping the team samples of the cats' white blood. After their LPL gene mutation was confirmed, Jones sent over a few of the cats.
They looked like ordinary cats unless you stared into their white retinas.
"The cat was a miracle because it was a naturally occurring model for the disease we had in humans," Kastelein said.
When the gene therapy worked on the cats they knew their discovery was ready for human trials.
III.
The team's research had reached what drug developers call the "valley of death" — the critical point where a university discovery is ready to make the leap to human tests and then into commercial development.
It's when investors and business experts step in to raise money for the clinical trials and the manufacturing and licensing costs.
"The moment that happens, we at universities lose control," Hayden said.
But it's the only way scientific discoveries ever get to patients, because universities don't make drugs.
For his birthday, he travelled back to where he came from in Spain, had two beers, and he was dead three days later. And the boy just wanted to have fun just one time in his life — and he paid with his life.
By 1998, Kastelein had returned to the Netherlands to continue the research by studying LPLD in European patients.
He became a world expert in lipid disorders and witnessed many tragic examples of what can happen when patients fail to follow their dietary restrictions, especially when it comes to alcohol.
"There's an absolute prohibition of alcohol for these people," Kastelein said, recalling one teenager who wanted to celebrate his 18th birthday.
"For his birthday, he travelled back to where he came from in Spain, had two beers, and he was dead three days later. And the boy just wanted to have fun just one time in his life — and he paid with his life."
Kastelein was determined to get the UBC discovery to patients. He joined with some fellow scientists at the University of Amsterdam to form a drug company called Amsterdam Molecular Therapeutics (AMT).
Back at UBC, Hayden's team continued to offer laboratory support to the new Dutch company.
"We took over all the science — the mice science, the cat science, the virus science," Kastelein said.
The next step was to convince the European health authorities to approve the first human trials.
We had to go through all sorts of regulations so that this virus wouldn’t escape into the environment and cause walking zombies or something.
It would be one of the first times human DNA, packaged in a virus, was ever injected into human volunteers. And the health regulators were extremely cautious.
"We had to go through all sorts of regulations so that this virus wouldn’t escape into the environment and cause walking zombies or something," Kastelein said.
The company fought "round after round of bureaucracy" before being granted permission, he said.
In the summer of 2005, the team enrolled eight Dutch patients with LPLD and followed strict environmental containment procedures for the trial.
"We all were completely masked up, suited up," Kastelein said. "The patients, when they had to urinate, that was biohazardous material that had to be destroyed. They had to be kept in isolation, so the air was not allowed to escape from the room.
"We now understand that the viruses are totally harmless, that all that suiting up was totally nonsense. We don’t do that anymore, but in those days it was really something."
The treatment involved up to 60 injections into a patient's leg muscles — all in a single session.
Almost immediately, the levels of fat in the patients' blood appeared to drop.
The first phase of human testing was successful, so the company decided to move to a second phase — and that's when the Glybera story returned to Canada.

One of the largest groups of LPLD patients in the world is clustered along Quebec's Saguenay River. There is a special lipid clinic in Chicoutimi, where doctors treat those suffering from LPLD.
That's how Cynthia Turcotte first heard about the clinical trial for Glybera in 2008. When her Montreal doctor mentioned the experimental therapy being tested a few hours away, she jumped at the chance to volunteer.
"When I heard about Glybera and the possibility to help me have a better life and maybe have children more easily, I said, 'Yeah, I want to go there. I want it."
She had 45 injections into her leg muscles. The one-time treatment was over in a few hours but its effectiveness lasts to this day.
And because of Glybera, she was able to give birth to two healthy children.
"For me, it was like a miracle. I didn't believe that I would have children. And they still are my two miracles."
But she would be one of the few people in the world to ever receive the life-changing treatment.
IV.
Back in the Netherlands, AMT had a facility ready to start manufacturing Glybera. But company officials were still fighting to convince the European Medicines Agency (EMA) to approve the drug for sale in Europe.
LPLD is rare, so the clinical trials were small. One of the EMA committees wanted more data before approving the company's application.
After the process stalled at the committee stage, AMT appealed directly to the politicians at the European Commission.
"There was a lot of fighting and politics, and it was very unpleasant," Kastelein said. "But in the end, it was a huge victory because it was the first registered gene therapy."
But in victory, there was also defeat.

In the 2 1/2 years it took to win EMA approval, AMT, which had no other products to sell and no revenue from Glybera, lost millions of dollars. The company was formally liquidated in 2012. Its assets were acquired by a new private company, uniQure.
Dr. Sander van Deventer is uniQure's current chief scientific officer, but he was also an executive with AMT, as it struggled to get EMA licence approval.
"These sorts of things have business consequences. We had lost all our financial capabilities," van Deventer said of AMT's demise.
To raise enough money to launch Glybera, uniQure partnered with an Italian pharmaceutical company, Chiesi Farmaceutici. For 31 million euros, Chiesi acquired the rights to sell Glybera in Europe, while uniQure retained the rights to the Canadian and U.S. markets.
"There was a totally new management," said Kastelein, whose only role in the new company was as a scientific consultant.
When Glybera finally went on sale in Europe in 2015, it made international headlines for its price: approximately $1 million US for a single dose.
Back in Canada, Hayden was embarrassed.
"I was not happy," he said. "I did not know what they were going to charge."
Hayden and UBC had no financial stake in Glybera.
"That's not the reason for doing this," he said. "The reason for doing this is to have some impact on patients."
But Hayden knew patients could not afford a $1-million drug.
"To be quite frank, this was not something I was particularly proud of, that the pricing of this made it out of the reach of the patients."
Kastelein was also shocked by the price, which he first heard about from a German colleague who wanted to prescribe the drug for a patient.
"The problem is, with people like me and Michael Hayden, we never have any say about pricing," he said. "By the time there's a pricing, we're gone already. We've done the science and the clinical work and everything, and then it's the commercial and financial people that determine the price."

The decision to price Glybera at $1 million was based on a business calculation, according to van Deventer.
To set a price, they compared Glybera to other drugs that treat rare diseases.
Because Glybera is a one-time treatment that can last at least 10 years (according to patient data collected so far), the $1-million price seemed reasonable, van Deventer said.
"It's not a crazy price," he said. "People say it's the most expensive drug in the world and what have you, but in the end, all of these products, even priced at $1 million, are going to be generally cheaper than replacement therapy."
Replacement therapy refers to drug treatments that replace missing proteins rather than repairing defective genes. Unlike Glybera, most replacement therapies must be given for the rest of a patient's life. Many of those drugs cost more than $300,000 per patient per year.
The price tag made it difficult to convince European governments and private insurance companies to pay for Glybera.
The world’s first gene therapy had only one customer: a German woman with LPLD who suffered such debilitating pancreatitis she had been hospitalized in intensive care more than 40 times.
Her doctor, Elisabeth Steinhagen-Thiessen, is also an expert in LPLD and had to fight to get an insurance company to pay for the dose.
It was worth it, she said.
"Everything went fine. She's back at work. And she's happy."
The woman has never had another pancreatitis attack.
In April 2017, just two years after it first went on the market, Chiesi announced it was abandoning Glybera. The company allowed the European marketing licence to expire.
Three doses left on the shelf were basically given away. A patient in Italy was treated for 1 euro, and two German patients also received doses for 1 euro each after Dr. Steinhagen-Thiessen asked Chiesi for the leftover product.
She said it worked for both her patients.
"The man we treated, I talked to him on the phone two weeks ago and he always says to me he feels like he’s newborn," she said.
Pricing shouldn't be a political decision. It should be a rational decision based on merits and values.
After giving away the three doses, Chiesi had nothing more to do with Glybera.
"All product rights were returned to uniQure, and Chiesi is no longer involved in the management of the product," Chiesi spokesperson Valentina Biagini said.
The drug was given to just 31 people in the entire world, most of whom were treated for free in clinical trials. UniQure has no plans to licence Glybera in Canada or the U.S.
Van Deventer says the company never considered lowering the price.
"Why would we? Pricing shouldn't be a political decision. It should be a rational decision based on merits and values," he said. "Hundreds of millions of investor money has gone into the company, and if there is no return for those investments, there will be no new drugs because nobody's going to do that in the future, right?"
The drug industry is accustomed to failure. Only a fraction of drugs in development ever make it to market. But unlike most experimental compounds that end up being too toxic or ineffective, Glybera actually worked. It is still preventing potentially deadly pancreatitis in the patients who were treated.
In a perfect world, van Deventer says, Glybera would still be on the market.
"It's a shame because it does work and I think people accept that it works and it should have been a product for sure."
But he said the costs for uniQure were too high.
"You need to maintain the factory, you need to do the paperwork, you need to test the product, you need to make new product batches all the time because product expires," he said.
"I’m really disappointed that after all the work, and also after having been very much involved with all these patients all that time, that this is the end. It took a long time for me to accept my loss, but eventually, after 15 years, I had to accept my defeat here."
V.

For Hayden and Kastelein, Glybera remains a career highlight.
"It's not the perfect ending that I would have liked, but life isn't always perfect," Hayden said. "Obviously this is disappointing. At the same time, it’s led to feasibility, proof of concept, for many other approaches that are coming down the line."
Kastelein remembers being angry when Glybera fizzled in the marketplace, but that feeling has long since passed.
"Because this has taken so long, it kind of wears you out in a way," he said. "So your anger kind of dissipates with time."
In the end, Glybera was defeated not by science but by the harsh reality of the drug business. It's not enough for a drug to be effective; it has to be profitable, too.
"It's kind of the law of nature," Kastelein said.
"If it's not commercially viable to produce a certain therapy, unfortunately, in our Western society, it does not happen."